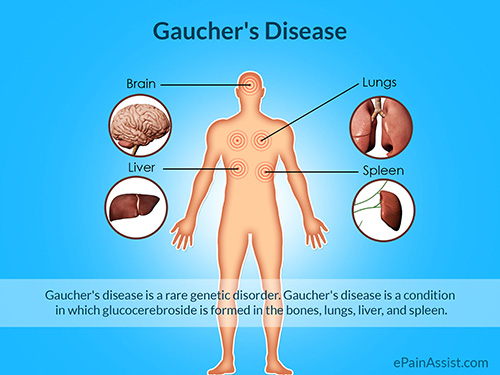
 Gaucher (or Gaucher’s) Disease is caused by a hereditary deficiency of the enzyme glucocerebrosidase. The enzyme acts on the fatty acid glucosylceramide. When the enzyme is defective or deficient, glucosylceramide can accumulate in the spleen, liver, kidneys, lungs, brain and bone marrow.
Gaucher (or Gaucher’s) Disease is caused by a hereditary deficiency of the enzyme glucocerebrosidase. The enzyme acts on the fatty acid glucosylceramide. When the enzyme is defective or deficient, glucosylceramide can accumulate in the spleen, liver, kidneys, lungs, brain and bone marrow.
Manifestations may include (often painless) enlarged spleen and liver, liver malfunction, skeletal disorders and bone lesions that may be painful, neurologic complications, swelling of lymph nodes and (occasionally) adjacent joints, distended abdomen, a brownish tint to the skin, anemia, low blood platelets and yellow fatty deposits on the white of the eye (sclera). Persons affected most seriously may also be more susceptible to infection.
 Around 1 in 100 people in the general U.S. population is a carrier for Type I Gaucher’s disease. The disease is caused by a recessive mutation in a gene located on chromosome 1 and affects both males and females. Among people of Ashkenazi Jewish descent, the rate of carriers is considerably higher, at roughly 1 in 15 people.
Around 1 in 100 people in the general U.S. population is a carrier for Type I Gaucher’s disease. The disease is caused by a recessive mutation in a gene located on chromosome 1 and affects both males and females. Among people of Ashkenazi Jewish descent, the rate of carriers is considerably higher, at roughly 1 in 15 people.
Some forms of Gaucher’s disease (including Type I) can be treated with enzyme replacement therapy (an IV therapy) to bolster the deficient or defective glucocerebrosidase. Enzyme replacement therapy has been shown to be effective when it is started before bone damage or other complications occur and is done regularly.
The disease is named after the French doctor Philippe Gaucher, who originally described it in the year 1882.


